ExomeNext® and ExomeReveal®
Maximize the Diagnostic Yield Over Standard Exomes
Since 2011, our ExomeNext test has provided high quality, comprehensive genomic analysis for patients affected by undiagnosed rare diseases and neurodevelopmental disorders.
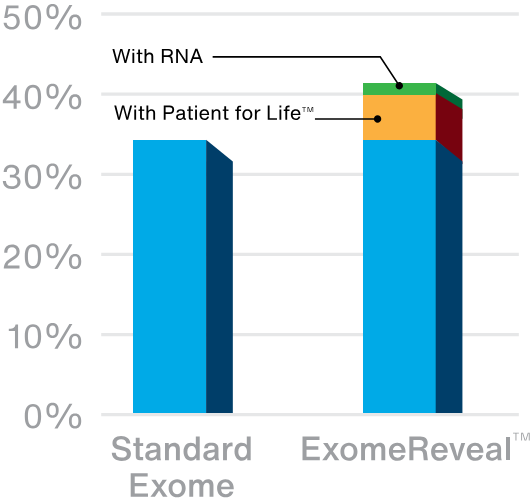
Exome sequencing has a diagnostic yield of up to ~35% depending on the indication for testing. We increase that diagnostic yield with our unique Patient for Life program, which offers continuous, lab-driven reanalysis. About 1 in 20 patients (5%) who initially have negative or uninformative results get new answers over time, mainly as a result of research that advances our understanding of gene-disease relationships.
Introducing ExomeReveal
Our latest innovation in exome testing, ExomeReveal, adds the power of RNA analysis to exome testing to unlock even more answers for families. Based on available data, RNA analysis increases the diagnostic yield by another 2-3%. When combined with the impact of Patient for Life, ExomeReveal has a 20% relative increase in diagnostic yield over standard exome tests.
| Test Code | Test Name | TAT[1] | Genes |
|---|---|---|---|
| Test Code: 9500 | Test Name: ExomeNext-Select™ |
TAT:
|
Genes: up to 500 SEE COVERAGE DETAILS |
| Test Code: 9999R | Test Name: ExomeNext-Rapid® |
TAT:
|
Genes: ~ 20,000 SEE COVERAGE DETAILS |
| Test Code: 9990 | Test Name: ExomeReveal™ |
TAT:
|
Genes: ~ 20,000 SEE COVERAGE DETAILS |
| Test Code: 9920 | Test Name: ACMG Secondary Findings |
TAT:
|
Genes: 84 |
| Test Code: 9900-M | Test Name: Mito DNA |
TAT:
|
Genes: 25 |
| Test Code: 9900 | Test Name: ExomeNext® (includes Patient Only Chart, Proband, and Relative orders) |
TAT:
|
Genes: ~ 20,000 SEE COVERAGE DETAILS |
|
Footnotes: [1] TAT is estimated based on typical lab performance and may vary depending on a number of factors, including but not limited to specimen sufficiency, order volume, and unforeseen circumstances.; [2] TAT is for trio exome orders with Blood EDTA specimens. TAT may be extended in rare instances when reruns are required. Non-trio orders and alternative specimen types may have extended TATs.; [3] ExomeReveal™ includes supplemental RNA analysis and is available with all ExomeNext® orders except ExomeNext-Rapid. RNA analysis requires an additional 3-4 weeks TAT for reporting after initial DNA sequencing results.; [4] Secondary findings: Check "decline" to opt-out of the ACMG Recomended List of secondary findings. If left unchecked, secondary findings will be reported.; [5] Only one test can be selected per single gene/condition order. |
|||
- AmbryPort Login or Create an Account
- Order a Sample Kit
We offer family variant testing at no additional cost
We offer family variant testing for all blood relatives of patients who undergo full single gene sequencing, multigene panel testing or exome sequencing at Ambry Genetics and are found to have a pathogenic or likely pathogenic variant. Testing must be completed within 90 days of the original report date. Whenever possible, more closely related relatives should be tested before more distant relatives. If you or a family member are interested in learning more about our family testing program or when family testing may be clinically indicated, please contact us or your provider for additional information. Note that Ambry can only provide such family testing services to patients receiving medical care in the U.S or US territories.
Order NowPatient for Life Program
For patients undergoing this test, Ambry will continually review data for potential pathogenic or likely pathogenic variants in newly characterized genes and will proactively issue reclassification reports, as applicable. Ask your Genomic Science Liaison for more details.
Why Is This Important?
Identifying an underlying genetic cause for rare diseases and neurodevelopmental disorders can clarify a diagnosis and inform recommendations for personalized medical management. Benefits can include:
- Tailoring medical care, with appropriate specialist referrals, screening and surveillance
- Guiding therapy selection and qualifying patients for clinical trials
- Informing family decisions, including reproductive counseling, childcare choices, and financial planning
- Creating opportunities for community and advocacy
When To Consider Testing
Exome sequencing is a comprehensive testing approach recommended for the evaluation of:
- Multiple congenital anomalies (MCA)
- Developmental delay (DD)
- Intellectual disability (ID)
- Autism spectrum disorder (ASD)
- Unexplained epilepsy
- Cerebral palsy (CP)
Test Description
Testing with ExomeNext and ExomeReveal includes whole exome sequencing of ~20,000 nuclear genes using next generation sequencing methods. Genetic variants are filtered through our in-house bioinformatics pipeline and analyzed by our medical team. Filtering is performed based on inheritance models and HPO terms are applied manually to protect limited evidence genes or to increase the diagnostic yield in cases where phenotype is limited. Variants are reviewed to determine pathogenicity and clinical correlation with the patient’s clinical symptoms. Relevant variants that meet quality thresholds are reported.
Exome testing can be ordered as a:
- Trio – Proband plus two close biological relatives, usually the parents
- Duo – Proband plus one close biological relative
- Proband only
Trio testing is recommended if relatives are available and consent to testing. Trio testing allows reporting of all ~20,000 genes, including relevant findings in uncharacterized genes. If a Proband or Duo is ordered, reporting will only include ~5,000 characterized genes.
If mitochondrial testing is selected, the mitochondrial genome is also analyzed for a defined list of established disease-causing variants.
Download Mitochondrial DNA Variant List
Exome testing includes analysis for secondary findings (SF) in the 81 genes recommended by American College of Medical Genetics and Genomics (ACMG) guidelines. These genes are related to conditions for which medical management is available to alter the course of the disease. Secondary findings are available for all members of the Duo/Trio and report separately for relatives. Patients can be opted out if preferred.
Download ACMG Secondary Findings List
When ExomeReveal is selected and a PAXgene tube blood sample is included, testing includes supplemental RNA analysis for qualified variants expected to impact splicing. RNA analysis reports 3-4 weeks after initial DNA test results.
Exome sequencing: Recommended by clinical guidelines
Many professional societies recommend exome sequencing as a first-line test, which can be ordered as soon as symptoms or features are identified.
The American Academy of Pediatrics (AAP) recommends exome/genome sequencing as first-tier testing for1:
- Global Developmental Delay
- Intellectual Disability
Read the updated 2025 guidance
The American College of Medical Genetics and Genomics (ACMG) recommends exome sequencing as a first-tier test for2:
- Developmental Delay
- Intellectual Disability
- Congenital Anomalies
The National Society of Genetic Counselors (NSGC) and American Epilepsy Society (AES) recommend exome or genome sequencing for individuals with3:
- Unexplained Epilepsy
References:
1Rodan LH, Stoler J, Chen E, et al. Genetic evaluation of the child with intellectual disability or global developmental delay: clinical report. Pediatrics. 2025;156(1):e2025072219. doi:10.1542/peds.2025-072219
2Manickam K, McClain MR, Demmer LA, et al. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23(11):2029–2037. doi:10.1038/s41436-021-01242-6
3Smith L, Malinowski J, Ceulemans S, et al. Genetic testing and counseling for the unexplained epilepsies: an evidence-based practice guideline of the National Society of Genetic Counselors. J Genet Couns. 2023;32(2):266–280. doi:10.1002/jgc4.1646