Exome Sequencing Overview For Contract Services
Exome sequencing is a robust technology that sequences all of the functionally relevant regions of essentially all 20,000 genes of the human genome and has the capacity to pinpoint rare genetic lesions in an unbiased and efficient way. Exome sequencing is a cost-effective and comprehensive method to rapidly detect the underlying etiology in patients with genetic disease.1-6
Exome Sequencing Processes

Reference 7,8
Bioinformatics
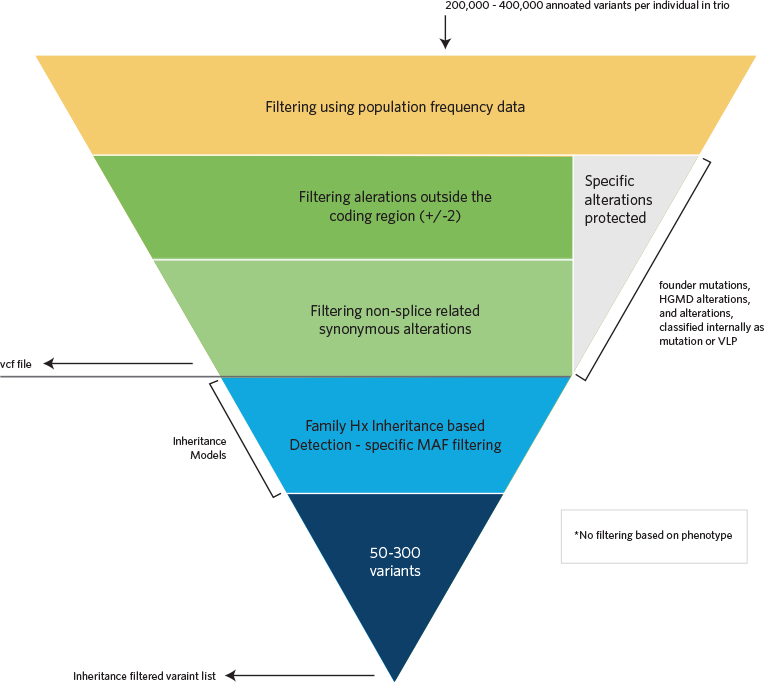
Sequence alignments to the reference human genome (GRCh37) and variant calls are generated using various bioinformatics tools (CASAVA, NovoAlign, Pindel, and GATK). The HGMD, the dbSNP database, the 1000 Genomes Project, HapMap data, and online search engines (e.g., PubMed), and Ambry's internal database are used to search for previously described gene mutations and polymorphisms. Data are annotated with the Ambry Variant Analyzer tool, including nucleotide and amino acid conservation, biochemical nature of amino acid substitutions, population frequency (Exome Variant Server https://geno2mp.gs.washington.edu/Geno2MP/, Exome Aggregation Consortium https://gnomad.broadinstitute.org/, and the 1000 Genomes Project), and predicted functional impact (including PolyPhen and SIFT in silico prediction tools). Sequence alignments of the reads are viewed using IGV (Integrative Genomics Viewer) software. Stepwise filtering includes the removal of common SNPs, intergenic and 3’/5’ UTR variants, non–splice related intronic variants, and synonymous variants (except those at the first and last nucleotide position of an exon). The filtering pipeline protects all variants annotated within the HGMD and/or the OMIM databases.
Inheritance Filters
Variants are then filtered further based on family history and possible inheritance models using the affected status of each family member whose exome was sequenced and compares the proband’s genotype of each detected alteration to the genotype of the family members (most commonly parents). Alterations survive the filtering if they are consistent with Mendelian inheritance models and the affected status of each family member. A minimum of four inheritance models are executed for each family (autosomal dominant (AD), autosomal recessive (AR), X-linked recessive, X-linked dominant, and Y-linked in male probands). For increased sensitivity of potential phenocopies and genes associated with reduced penetrance, autosomal and X-linked “proband-only” models are also generated for each proband that captured heterozygous, homozygous, compound heterozygous, and hemizygous alterations irrespective of cosegregation based on family members’ genotypes.
Computational Filtering
High Diagnostic Rate
Exome is 2-3 times more likely to identify the underlying genetic defect than traditional tests
- Reaching a diagnosis means an end to the expensive, time-consuming, and potentially invasive diagnostic odyssey that burdens patients, families, and the healthcare system.
- The diagnostic rate for WES ranges from 25-35% based on published literature.9-16
- This rate is two to three times as likely as traditional methods to identify the underlying cause of a patient’s genetic disease.2-5, 17
- More importantly, this rate is continually increasing due to the rapid rate of new gene-disease discoveries. 10, 18
References
- Tan TY, Dillon OJ, Stark Z, et al. Diagnostic Impact and Cost-effectiveness of Whole-Exome Sequencing for Ambulant Children With Suspected Monogenic Conditions. JAMA Pediatr. 2017.
- Vissers LE, van Nimwegen KJ, Schieving JH, et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genetics in medicine : official journal of the American College of Medical Genetics. 2017.
- Stark Z, Schofield D, Alam K, et al. Prospective comparison of the cost-effectiveness of clinical whole-exome sequencing with that of usual care overwhelmingly supports early use and reimbursement. Genetics in medicine : official journal of the American College of Medical Genetics. 2017;19(8):867-874.
- Gomez CM, Das S. Clinical exome sequencing: the new standard in genetic diagnosis. JAMA Neurol. 2014;71(10):1215-1216.
- Monroe GR, Frederix GW, Savelberg SM, et al. Effectiveness of whole-exome sequencing and costs of the traditional diagnostic trajectory in children with intellectual disability. Genetics in medicine : official journal of the American College of Medical Genetics. 2016;18(9):949-956.
- Soden SE, Saunders CJ, Willig LK, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Science translational medicine. 2014;6(265):265ra168.
- Voelkerding KV, et al. Next Generation Sequencing for Clinical Diagnostics-Principles and Application to Targeted Resequencing for Hypertrophic Cardiomyopathy. Journal of molecular Diagnostics. 2010;5(12): 539-551.
- Biesecker, LG and Green RC. Diagnostic Clinical Genome and Exome Sequencing. New England journal of medicine. 2014;370(25):2418.
- Farwell KD, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(7):578-586.
- Farwell Hagman KD, et al. Candidate-gene criteria for clinical reporting: diagnostic exome sequencing identifies altered candidate genes among 8% of patients with undiagnosed diseases. Genetics in medicine : official journal of the American College of Medical Genetics. 2017;19(2):224-235
- Yang Y, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. The New England journal of medicine. 2013;369(16):1502-1511.
- Yang Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312(18):1870-1879.
- Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA.2014;312(18):1880-1887.
- Srivastava S, et al. Clinical whole exome sequencing in child neurology practice. Annals of neurology. 2014;76(4):473-483.
- Iglesias A, et al. The usefulness of whole-exome sequencing in routine clinical practice. Genetics in medicine : official journal of the American College of Medical Genetics. 2014;16(12):922-31.
- Retterer K, et al. Clinical application of whole-exome sequencing across clinical indications. Genetics in medicine : official journal of the American College of Medical Genetics. 2016;18(7):696-704.
- Shashi V, et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genetics in medicine : official journal of the American College of Medical Genetics. 2013;16(2):176-82
- Boycott KM, et al. International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. American journal of human genetics. 2017;100(5):695-705.